


















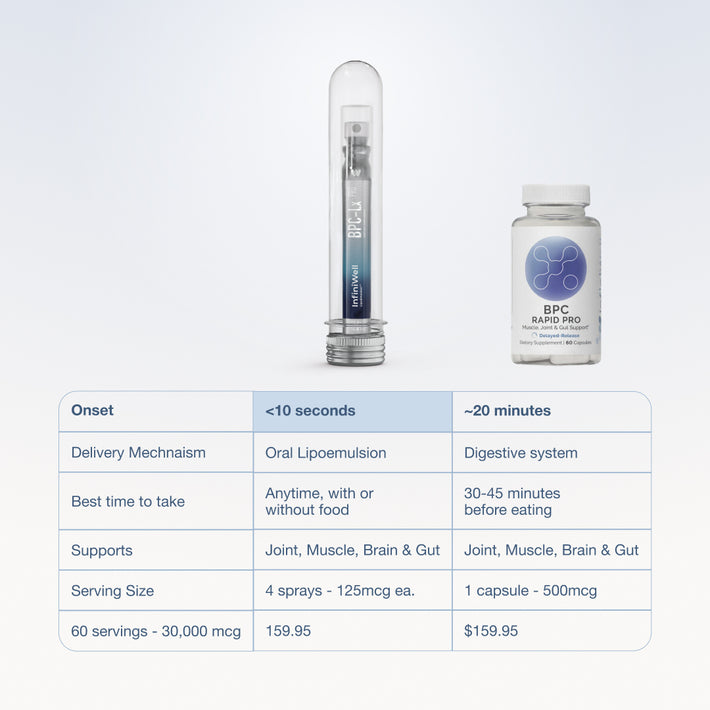


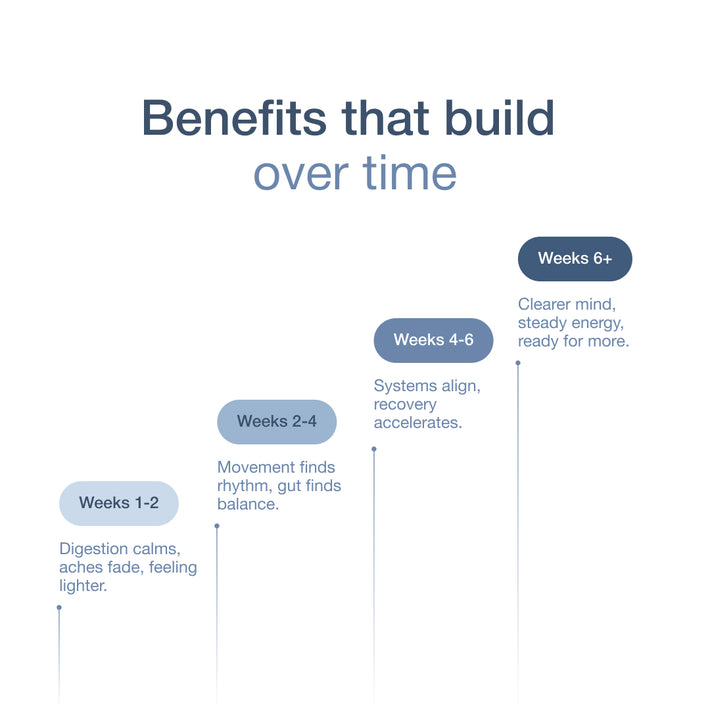





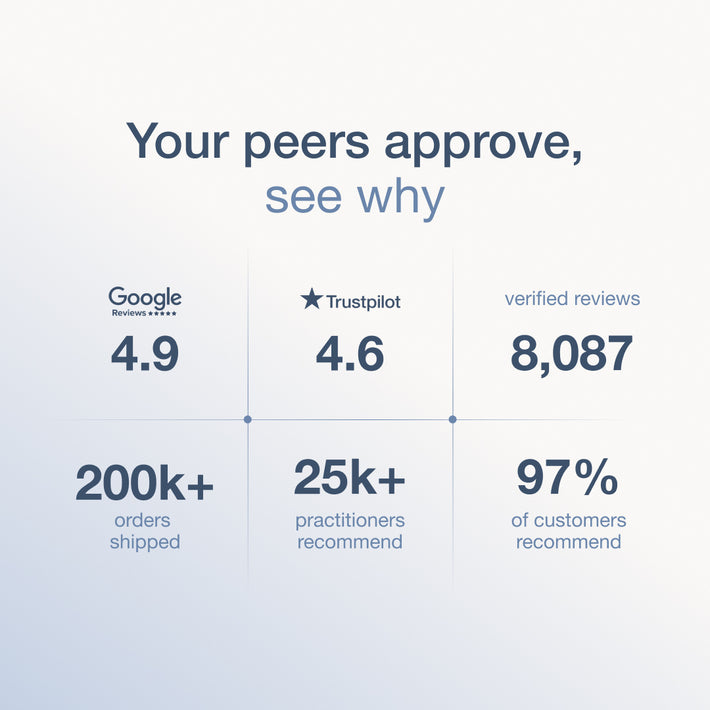





































Key takeaways
-
Regulators and industry leaders are revisiting what qualifies as a dietary ingredient, a question that has shaped the market for decades.
-
Probiotics remain in a gray area, with science advancing faster than the current regulatory framework.
-
Ingredient identity is becoming more technical, especially for proteins and microbial strains, where small differences can be significant.
-
Delivery systems are gaining attention, as they can influence how and where ingredients function in the body.
-
New production methods like fermentation and cell culture are pushing regulators to reconsider how ingredients are evaluated and approved.
On March 27, 2026, the Food and Drug Administration held a public meeting that put dietary supplement regulations under a serious spotlight.
The agenda covered three main topics:
-
What legally counts as a dietary ingredient
-
How new production methods fit into that picture
-
What science should guide how complex ingredients like proteins, enzymes, and probiotics are defined
The conversation was long overdue, and the outcomes will be important for anyone who takes supplements or works in the industry.
Why this FDA meeting is significant
The FDA does not hold public meetings like this one very often. March 27, 2026 was one of those rare occasions where regulators, scientists, and industry leaders sat in the same room to address a legal question that has shaped the supplement market for over three decades: What actually counts as a dietary ingredient under U.S. law?
The guest list reflected how seriously this was taken:
-
Discussion leads: Dr. Cara Welch (FDA Office of Dietary Supplement Programs), Dan Fabricant (Natural Products Association), Jensen Jose (Center for Science in the Public Interest)
-
Additional perspectives: University of Illinois, Novonesis, Council for Responsible Nutrition, International Probiotics Association
The whole conversation traced back to a single phrase inside DSHEA, the Dietary Supplement Health and Education Act, passed in 1994: "a dietary substance for use by man to supplement the diet." Thirteen words that the FDA and the supplement industry have been interpreting differently for years.
As NutraIngredients.com reported, March 27 was the first formal effort to resolve that disagreement in over 30 years.
What counts as a dietary ingredient under U.S. law
DSHEA was passed in 1994 with language intended to leave room for new ingredients to enter the market, even if they had no prior history in the conventional food supply. Over time, the FDA began interpreting that section more narrowly, requiring a traceable connection to existing human food before a substance could qualify as a dietary ingredient.
Jensen Jose argued that grounding dietary substances in the food supply gives regulators and consumers a reliable safety baseline. Dan Fabricant pushed back, pointing to DSHEA's legislative history and making the case that the law was written to allow ingredients like CoQ10, melatonin, and probiotics to exist in the market, even before they had a clear food supply connection.
As a result of the FDA's current reading, truly new ingredients face a harder road to market, even when the science behind them is solid. Industry leaders at the meeting consistently argued that the law was always meant to grow alongside science, not hold it back.
Why probiotics still have no clear regulatory category
Probiotics are one of the most popular dietary supplements available, yet they sit in a genuinely unclear regulatory space. That gap was a major talking point at the March 27 meeting, and for good reason.
The issue goes back to timing. Probiotics were not formally defined until 2001, which was seven years after DSHEA was written. Because they were left out of the original law, they have never been officially recognized as a dietary ingredient category. Many probiotic strains end up going through the GRAS pathway, which was built for food additives, not dietary supplements.
Amy Smith of Kerry Group and president of the International Probiotics Association argued that exposure to microbes through the human diet, whether intentional or incidental, should factor into how the FDA evaluates these ingredients. Andrea Wong of the Council for Responsible Nutrition (CRN) added that DSHEA was always meant to be broad and was never intended to exclude substances not already found in food.
The science on probiotic safety is well established. The regulations just have not kept up.
How supplement ingredients are identified and tested
Establishing what is actually in a supplement, and confirming it matches the label, is more involved than most people expect. This part of the meeting looked at what it really takes to verify ingredient identity as dietary ingredients get more complex.
Professor Gonzalez de Mejia from the University of Illinois explained that for protein-based ingredients, identity is not just about composition. The amino acid sequence, how the protein is structured, and how it folds all count. A single amino acid difference can change how a substance behaves in the body and, in some cases, affect safety.
Frank Romanski illustrated this with undenatured collagen type II, noting that its effectiveness depends on preserving a very specific and small fraction of undenatured collagen. Only validated ELISA testing can confirm it is present in the right form.
Products tested through other methods may carry the same label claims but are not the same thing scientifically. That gap raises concerns around both quality and compliance.
Current good manufacturing practice standards require dietary supplement manufacturers to verify ingredient identity as part of the manufacturing process, but those standards will need to keep pace with how ingredients are evolving.
Why delivery affects whether a supplement works
Having a well-tested ingredient is only part of the equation. Whether it actually reaches the right place in the body is just as important, and this was one of the more practical discussions of the day.
Frank Romanski and other formulation experts made clear that for enzymes, probiotics, and peptides, delivery is not a bonus feature. It determines whether the ingredient gets to the right part of the gastrointestinal tract and actually does what it was made to do. Without the right delivery system, even a well-characterized ingredient may not produce the actual results it was designed for.
Advanced encapsulation systems, including capsule-in-capsule formats, were shown to make a real difference in delivery compared to standard formulations.
This raised a new regulatory question: If delivery determines how an ingredient functions, should it be considered part of that ingredient's identity?
Although the meeting did not resolve it, it put it firmly on the table.
How new production methods are changing supplement ingredients
The supplements on shelves today look very different from those available when DSHEA was written in 1994.
Precision fermentation, cell culture technology, and recombinant production are now being used to produce dietary ingredients at scale, and the FDA raised a direct question at the meeting: When a production method changes, does the ingredient change enough to need a fresh review?
Linda Nechmer of Novonesis described fermentation-derived ingredients as fully documented substances with clear sequences, structures, functions, and manufacturing histories. These are not experimental compounds. They are produced through validated processes with thorough analytical testing and have a track record across food, supplements, and healthcare that spans decades.
The gap is between what science can now define clearly and what the current regulatory system is set up to handle.
As noted by the FDA's Office of Dietary Supplement Programs, the agency is actively gathering input on how to evaluate ingredients made through precision fermentation, cell culture, and recombinant production going forward.
Why probiotic strain classification is important
Microbial ingredients like probiotics are not defined by a single data point. Gregory Leyer and other scientists at the meeting explained that microorganisms are classified at three levels:
-
Genus
-
Species
-
Strain
Each level means something different, and that distinction carries weight both scientifically and regulatorily.
Clinical outcomes for probiotics are almost always tied to a specific strain. Two strains within the same species can produce completely different results in the body. Safety, on the other hand, is generally assessed at the species level, using historical use in the human diet as the reference point.
The problem is that current FDA regulations do not consistently reflect this. Requiring strain-level presence in the food supply sets a bar that many well-supported strains cannot clear, even when the science on their safety and intended use is strong. This is one of the clearer points where the regulations and the science are out of step.
How new dietary ingredients get to market
The New Dietary Ingredient Notification process is the main pathway dietary supplement manufacturers use to introduce new dietary ingredients to the U.S. market. It requires companies to submit safety data and scientific evidence to the FDA before a new ingredient goes on sale, giving the agency a chance to review it before it reaches consumers.
When the process works as intended, it also rewards companies that invest in proper documentation. Compliant manufacturers get a level of regulatory recognition that distinguishes their products from those that skipped the process.
The issue raised at the meeting is that enforcement has been inconsistent, and that creates a problem for the whole industry.
Dan Fabricant and others argued that stronger FDA actions in this space go beyond holding companies accountable. They help set a quality floor that protects consumers and gives responsible dietary supplement manufacturers a reason to keep investing in proper testing, documentation, and identity verification.
The resource gap behind supplement regulation
One of the more candid conversations of the day was about FDA capacity. The Office of Dietary Supplement Programs reviews New Dietary Ingredient (NDI) notifications under tight staffing and timelines. The 75-day review window puts pressure on both the agency and the manufacturers submitting data, and that pressure grows as ingredients get more complex.
As more dietary ingredients come from precision fermentation, cell culture, and recombinant processes, the depth of what needs to be reviewed grows, too. Multiple speakers flagged this as a structural issue that will affect how quickly any changes from this meeting can actually be put into practice.
The Department of Health and Human Services oversees the FDA, and addressing capacity will need to be part of any real effort to update dietary supplement regulations. Expanding what qualifies as a dietary ingredient without expanding the ability to review those ingredients would create more delay, not better oversight.
What comes next for dietary supplement compliance
The public comment docket for this meeting stays open until April 27, 2026. This is a direct opportunity for manufacturers, researchers, and the public to weigh in on how the FDA defines "dietary substance" going forward. Comments can be submitted at regulations.gov, docket number FDA-2026-N-2047.
As Loren Israelsen of United Natural Products Alliance (UNPA) noted, the more comments the FDA receives, the stronger the push for meaningful change. Engaging now, before the comment period closes, is one of the most direct ways to influence where these regulations land.
Reviewing your supplement standards and what to check
As the regulatory picture develops, here are the areas worth paying attention to when reviewing supplement quality and compliance:
-
Verify full ingredient identity on product labels, including strain-level names for any probiotic products
-
Look for brands that publish batch-specific Certificates of Analysis covering finished batch testing results
-
Confirm that labeling clearly states the intended use and identity of each ingredient
-
Prioritize products from manufacturers following current good manufacturing practice standards with proper inspection and documentation in place
-
Review health claims carefully and check that they are backed by scientific evidence
-
Stay updated on upcoming FDA actions and any formal guidance that follows this meeting
What better regulations mean for the supplement industry
The supplement industry has grown well past vitamins and capsules. Fermentation-derived bioactives, precision amino acids, clinically studied microbial strains, and advanced delivery systems are now part of everyday supplement formulation. The law that governs them was written before most of these developments existed.
The FDA oversees:
-
How dietary ingredients are identified and tested
-
What types of health claims dietary supplements are allowed to make
-
What standards manufacturers and distributors must comply with across the manufacturing and distribution process
Regulatory action follows when science and regulations fall out of alignment.
What the March 27 meeting showed is that the FDA is ready to engage on these issues and is open to input from across the industry. Better defined regulations lead to more consistent quality, more accurate labeling, and a supplement market where scientific evidence and manufacturing standards are the expected baseline.
Visit the official FDA meeting page for a full overview.
FDA dietary supplement regulations: FAQs
-
SOURCES
You May Also Like
View All Articles

Why InfiniWell Costs More
InfiniWell’s price reflects how the products are sourced, formulated, tested, and manufactured before they ever reach the bottle. This breakdown shows where that cost comes from and what to look for when comparing supplement quality.